
Lax FDA Oversight of Medical Devices Exposed in Lawsuits
/By Fred Schulte and Holly K. Hacker, KFF Health News
Living with diabetes, Carlton “PeeWee” Gautney Jr. relied on a digital device about the size of a deck of playing cards to pump insulin into his bloodstream.
The pump, manufactured by device maker Medtronic, connected plastic tubing to an insulin reservoir, which Gautney set to release doses of the vital hormone over the course of the day. Gautney, a motorcycle enthusiast, worked as a dispatcher with the police department in Opp, Alabama.
The 59-year-old died suddenly on May 17, 2020, because — his family believes — the pump malfunctioned and delivered a fatal overdose of insulin.
“There’s a big hole left where he was,” said Gautney’s daughter, Carla Wiggins, who is suing the manufacturer. “A big part of me is missing.”
The wrongful-death lawsuit alleges the pump was “defective and unreasonably dangerous.” Medtronic has denied the pump caused Gautney’s death and filed a court motion for summary judgment, which is pending.
The pump Gautney depended on was among more than 400,000 Medtronic devices recalled, starting in November 2019, after the company said in a recall notice that damage to a retainer ring on the pump could “lead to an over or under delivery of insulin,” which could “be life threatening or may result in death.”

CARLA WIGGINS AND CARLTON GAUTNEY
As the recall played out, federal regulators discovered that Medtronic had delayed acting — and warning patients of possible hazards with the pumps — despite amassing tens of thousands of complaints about the rings, government records show.
Over the past year, KFF Health News has investigated medical device malfunctions including:
Artificial knees manufactured by a Gainesville, Florida, company that remained on the market for more than 15 years despite packaging issues that the company said could have caused more than 140,000 of the implants to wear out prematurely.
Metal hip implants that snapped in two inside patients who said in lawsuits that they required urgent surgery.
Last-resort heart pumps that FDA records state may have caused or contributed to thousands of patient deaths.
And even a dental device, used on patients without FDA review, that lawsuits alleged has caused catastrophic harm to teeth and jawbones. CBS News co-reported and aired TV stories about the hip and dental devices.
The investigation has found that most medical devices, including many implants, are now cleared for sale by the FDA without tests for safety or effectiveness. Instead, manufacturers must simply show they have “substantial equivalence” to a product already in the marketplace — an approval process some experts view as vastly overused and fraught with risks.
“Patients believe they are getting an implant that’s been proven safe,” said Joshua Sharlin, a former FDA official who now is a consultant and expert witness in drug and medical device regulation. “No, it hasn’t,” Sharlin said.
And once those devices reach the marketplace, the FDA struggles to track malfunctions, including deaths and injuries — while injured patients face legal barriers trying to hold manufacturers accountable for product defects.
In a statement to KFF Health News, the FDA said it “has a scientifically rigorous process to evaluate the safety and effectiveness of medical devices.”
‘Too Little, Too Late’
The FDA approved the MiniMed 670G insulin pump on Sept. 28, 2016, after its most stringent safety review, a little-used process known as premarket approval.
In a news release that day, Jeffrey Shuren, who directs the FDA’s Center for Devices and Radiological Health, lauded the device as a “first-of-its-kind technology” that would give patients “greater freedom to live their lives” and to monitor and dispense insulin as needed. The pump was tested on 123 patients in a clinical trial over several months with “no serious adverse events,” the release said. Shuren declined to be interviewed for this article.
The FDA’s enthusiasm didn’t last. In November 2019, Medtronic, citing the ring problem, launched an “urgent medical device recall” of the pumps, which it expanded in late 2021.
During an inspection at Medtronic’s plant in Northridge, California, FDA officials learned the company had logged more than 74,000 ring complaints between 2016 and the November 2019 recall.
More than 800 complaints weren’t investigated at all, according to the FDA, which sharply criticized the company in a December 2021 warning letter.
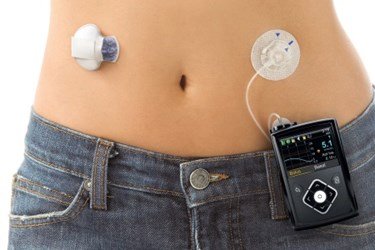
MiniMed 670G insulin pump (MEDTRONIC IMAGE)
Medtronic is facing more than 60 lawsuits filed by injured patients and their families and the company believes it may be hit with claims for damages from thousands more patients, the company disclosed in an August Securities and Exchange Commission filing.
Medtronic pumps that allegedly dispensed too much, or too little, insulin have been blamed for contributing to at least a dozen patient deaths, according to lawsuits filed since 2019. Some cases have been settled under confidential terms, while others are pending or have been dismissed. Medtronic has denied any responsibility in response to the lawsuits.
In one pending case, a Las Vegas man using the pump allegedly fell into an “insulin-induced coma” that led to his death in 2020. In another 2020 case, a 67-year-old New Jersey resident collapsed at her home, dying later the same day at a local hospital.
The recall notice Medtronic sent to a 43-year-old Missouri man’s home arrived a few days after police found him dead on his bedroom floor, his family alleged in a lawsuit filed in August. “Simply too little, too late,” the suit reads. The case is pending, and Medtronic has yet to file an answer in court.
Medtronic declined to answer written questions from KFF Health News about the pumps and court cases. In an emailed statement, the company said it replaced pump rings with new ones “redesigned to reduce the risk of damage” and “fulfilled all pump replacement requests at no cost to customers.”
In April, Medtronic announced that the FDA had lifted the warning letter a few days after it approved a new version of the MiniMed pump system.
Shortcut to Market
The 1976 federal law that mandated safety testing for high-risk medical devices also created a far easier — and less costly — pathway to the marketplace. This process, known as a 510(k) clearance, requires manufacturers to show a new device they plan to sell has “substantial equivalence” to one already on the market, even if the prior product has been recalled.
Critics have worried for years that the 510(k)-approval scenario is too industry-friendly to protect patients from harm.
In July 2011, an Institute of Medicine report concluded that 510(k) was “not intended to evaluate the safety and effectiveness of medical devices” and said “a move away from the 510(k) clearance process should occur as soon as reasonably possible.”
More than a decade later, that hasn’t happened, even amid mounting controversy over the clearance of hundreds of devices that employ artificial intelligence.
The FDA now clears about 3,000 low- to moderate-risk devices every year through 510(k) review, which costs the device maker a standard FDA fee of about $22,000. That compares with about 30 approvals a year through the stricter premarketing requirements, which cost nearly $500,000 per device, according to FDA data.
Diana Zuckerman, president of the National Center for Health Research, said even many doctors don’t realize devices cleared for sale typically have not undergone clinical trials to establish their safety.
“Doctors are shocked to learn this,” she said. “Patients aren’t going to know it when their doctors don’t.”
In response to written questions from KFF Health News, the FDA said it “continues to believe in the merits of the 510(k) program and will continue to work to identify program improvements that strengthen the safety and effectiveness of 510(k) cleared devices.”
The FDA keeps a tight lid on data showing which devices manufacturers choose to demonstrate substantial equivalence — what the agency refers to as “predicate” devices.
“We can’t get detailed data,” said Sandra Rothenberg, a researcher at the Rochester Institute of Technology. “It’s very hard for researchers to determine the basis on which substantial equivalence is being made and to analyze if there are problems.”
Rothenberg cited the history of “metal-on-metal” artificial hip implants, which under 510(k) spawned many new brands — along with a disastrous toll of patient injuries. The implants could release metal particles that damaged bone and led to premature removal and replacement, a painful operation. Just four of these hip devices have been the target of more than 25,000 lawsuits seeking damages, court records show.
In early 2016, the FDA issued an order requiring safety testing before approving new metal-on-metal hip devices.
Alarm Bells
Two former Medtronic sales executives in California argue in a whistleblower lawsuit that the 510(k) process can be abused. According to the whistleblowers, the FDA approved the Puritan Bennett 980, or PB 980, ventilator in 2014 based on the assertion it was substantially equivalent to the PB 840, an earlier mechanical ventilator long viewed as the workhorse of the industry.
Medtronic’s subsidiary company Covidien made its claim even though the device has completely different “guts” and operates using software and other “substantially different” mechanisms, according to the whistleblowers’ suit. In response, Medtronic said it “believes the allegations are without merit and has moved to dismiss the case.” The case is pending.
The whistleblowers argue the PB 980 ventilator was plagued by dangerous malfunctions for years before its recall in late 2021. One ventilator billowed smoke in an intensive care unit while the whistleblowers were told by one hospital that “the wheels for the ventilator cart may actually fall off the ventilator during transport,” according to the suit.
Batteries could die without warning, kicking off a scramble to keep patients alive; monitor screens froze up repeatedly or otherwise went on the blink; and, in several cases, alarm bells warning of a patient emergency rang continuously and could be quieted only by unplugging the unit from the wall socket and pulling out its batteries, according to the suit.
The December 2021 recall of the PB 980 cited a “manufacturing assembly error” that the company said may cause the ventilator to become “inoperable.”
Medtronic said in an email that the ventilator “has helped thousands of patients around the world,” including playing a “critical role in the global response to the COVID-19 pandemic.”
Late Warnings
The FDA operates a massive database, called MAUDE, to alert regulators and the public to emerging device dangers. The FDA requires manufacturers to advise the agency when they learn their device may have caused or contributed to a death or serious injury, or malfunctioned in a way that might recur and cause harm. These reports must be submitted within 30 days unless a special exemption is granted.
But FDA officials acknowledge that many serious adverse events go unreported — just how many is anybody’s guess.
Since 2010, the FDA has cited companies more than 5,000 times for not handling, reviewing, or investigating complaints properly, or for not reporting adverse events on time. For instance, the FDA cited an Ohio company that made electric beds and other devices more than 15 times for failing to properly scrutinize complaints or report adverse events, including the death of a patient who allegedly became trapped between a bedrail and mattress, agency records show.
In about 10% of reports, more than a year or two elapsed from when a death or serious injury occurred and when the FDA received the reports, a KFF Health News analysis found. That works out to nearly 60,000 delayed reports a year.
Experts and lawmakers say the FDA needs to find a way to detect safety problems quicker.
Sens. Chuck Grassley (R-Iowa) and Elizabeth Warren (D-Mass.) have tried for years to persuade the agency to add unique device identifiers to Medicare payment claim forms to help track products that fail. In an email statement to KFF Health News, Grassley called that a “commonsense step we can take up front to mitigate risk, improve certainty and save money later.”
The FDA said it is working to “strike the right balance between assuring safety and fostering device innovation and patient access.” Yet it noted: “Additional resources are required to establish a fully functioning active surveillance system for medical devices.”
For now, injured patients suing device companies often cite the volume of adverse event reports to MAUDE, or FDA citations for failing to report them, to bolster claims that the company knew about product malfunctions but failed to correct them.
In one case, a New York man is suing manufacturer Boston Scientific, claiming injuries from a device called the AMS 800 that is used to treat stress urinary incontinence.
Though Boston Scientific says on its website that 200,000 men have been treated successfully, the lawsuit argues complaints piled up in MAUDE year after year and no action was taken — by the company or by regulators.
The number of complaints filed soared from six in 2016 to 2,753 in 2019, according to the suit. By far, the largest category involved incontinence, the condition the device was supposed to fix, according to the suit. Boston Scientific did not respond to a request for comment. The company has filed a motion to dismiss the case, which is pending.
By the FDA’s own count, more than 57,000 of some 74,000 complaints Medtronic received about the MiniMed insulin pump’s retainer rings were reported to the agency. The FDA said the complaints “were part of the information that led to the compliance actions.” The agency said it “approved design and manufacturing changes to the retainer ring to correct this issue” and “has reviewed information confirming the effectiveness of the modification.”
“What is the threshold for the FDA to step in and do something?” said Mara Schwartz, who is a nurse, diabetes educator, and pump user. “How many deaths or adverse events does there have to be?”
In 2020, she sued Medtronic, alleging she suffered seizures when the pump mistakenly delivered an overdose of insulin. Medtronic denied her claims, and the case has since been settled under confidential terms.

Private Eyes
Some countries don’t trust the device industry to play such a key role in oversight.
Australia and about a dozen other nations maintain registries that measure the performance of medical devices against competitors, with an eye toward not paying for care for a substandard device.
That’s not likely to happen in the United States, where no device or drug manufacturer must demonstrate its new product is better than what’s already for sale.
Product liability lawsuits in the U.S. often cite troubling findings from overseas. For instance, registries in Australia and other countries pinpointed durability problems with the Optetrak knee implants manufactured by Florida device company Exactech years before a major recall. Exactech has declined comment.
The Australian surveillance network also detected deficiencies with the Medtronic PB 980 ventilator, prompting the country’s health authority to suspend its use for six months until Medtronic completed training for health care workers and took other steps to improve it, court records show. Medtronic told KFF Health News that it had “worked closely” with the Australian group to resolve the problems. “We take patient safety very seriously and have processes to identify quality issues and determine appropriate actions,” Medtronic said.
Registries have gained some traction in America. But so far, they typically have been controlled, and sometimes funded, by industry and medical specialty groups that share their findings only with doctors.
One private registry managed by the Society of Thoracic Surgeons, called Intermacs, tracks death and injury rates at 180 hospitals in the United States certified to implant a mechanical heart pump known as an LVAD. Some patients might find that information helpful, but it’s not available to them.
‘New and Exciting Features’
While the FDA clears thousands of devices for use based on the “substantial equivalence” premise, manufacturers often tout “new and exciting features” in their advertising and other marketing, said Alexander Everhart, a researcher at the Washington University School of Medicine in St. Louis.
These marketing campaigns have long been controversial, especially when they rely partly on wining and dining surgeons and other medical professionals to gain new business, or when surgeons have financial ties to manufacturers whose products they use. Orthopedic device makers have funneled billions of dollars to surgeons, including fees for consulting, doing medical research, or royalties for their role in fine-tuning surgical tools and techniques, even promoting the products to their peers.
Marketing campaigns directed at prospective patients may receive little scrutiny. The FDA has “limited resources to actively monitor the volume of direct-to-consumer advertising,” according to a Government Accountability Office report issued in September. From 2018 to 2022, the FDA took 255 enforcement actions involving advertising claims made for devices, according to the GAO report.
While manufacturers can advertise devices directly to patients, courts may not hold them accountable for communicating possible risks to patients.
Consider the case of Richard Greisberg, a retired electronics business owner in New Jersey. He sued Boston Scientific in 2019, years after having a Greenfield vena cava filter implanted. The device is intended to prevent blood clots that develop in the lower body from traveling into the lungs, which can be deadly.
Greisberg argued that the device had migrated in his body, causing pain and other symptoms and damage that took years to identify. Representing himself in court, he tried to argue that nobody had told him that could happen and that if they had done so he wouldn’t have agreed to the procedure.
He lost when the judge cited a legal doctrine called “learned intermediary.” The doctrine, which is recognized in many states, holds that manufacturers must warn only physicians, who are presumed to have the knowledge to understand a medical device’s risks and relay them to patients.
The court ruled that a 27-page manual the manufacturer sent to the physician who implanted it, which included details about possible risks, was adequate and tossed the case.
Greisberg, 81, felt sucker-punched. “They never gave me any warning about what could happen down the road,” he said in an interview. “I never had a chance to have my day in court.”
The family of PeeWee Gautney also faces challenges pursuing the insulin pump lawsuit.
Gautney died in a motel room in Destin, Florida, a day after riding his Harley-Davidson to the Panhandle beach town on a weekend jaunt. The MiniMed pump was still strapped to his body, according to a police report.
Medtronic had sent Gautney a form letter in late March 2020, less than two months before he died, advising him to make sure the ring was locking in place correctly. A week later, he wrote back, telling the company: “It’s fine right now,” court records show.
Wiggins, 33, his daughter, who is also a neonatal respiratory therapist, said she believes a crack in the retainer ring caused it to release too much insulin, which her dad may not have recognized.
“It should never be put on the patient to determine if there is a problem,” Wiggins said.
Medtronic has denied the pump failed and caused Gautney’s death. The FDA approved the device knowing patients faced the risk of it administering wrong doses, but believed the benefits outweighed these risks, Medtronic argued in a motion for summary judgment in September. The motion is pending.
Medtronic also cited a legal doctrine holding that Congress granted the FDA sole oversight authority over devices receiving premarket approval, which preempts any product defect claims brought under state laws. Manufacturers have drawn on the preemption defense to sidestep liability for patient injuries, and often win dismissal, though federal courts are split in applying the doctrine.
Wiggins hopes to beat those odds, arguing that the December 2021 FDA warning letter reveals that Medtronic violated safety and manufacturing standards.
Her lawyer, Scott Murphy, said that insulin pumps are “really wonderful” devices for people with diabetes when they work right. He argues that the FDA records confirm that Medtronic significantly downplayed its pump’s hazards.
“The risks get minimized and the benefits exaggerated,” he said.
KFF Health News is a national newsroom that produces in-depth journalism about health issues.




